In this condensed tutorial, Vessel presents three real-world cases of patients with confirmed progressive disease in which liquid biopsy was used to identify actionable targets. The tutorial also includes a brief summary of the patient's history and the results of the liquid biopsy.
In this condensed tutorial, we want to walk you through real-world cases as presented in ourMolecular Cancer review (original Figure 3 from the review). The cases here represent scenarios in which the goal was to identify actionable targets from plasma. In the next set of case studies to be launched, we will walk through the other 3 cases (original Figure 4 in the review), which were scenarios in which we performed serial monitoring via shallow whole-genome sequencing (sWGS) alone to follow the patient’s clinical course.
Getting started
Let’s start with Case 1 in Figure 1 below.
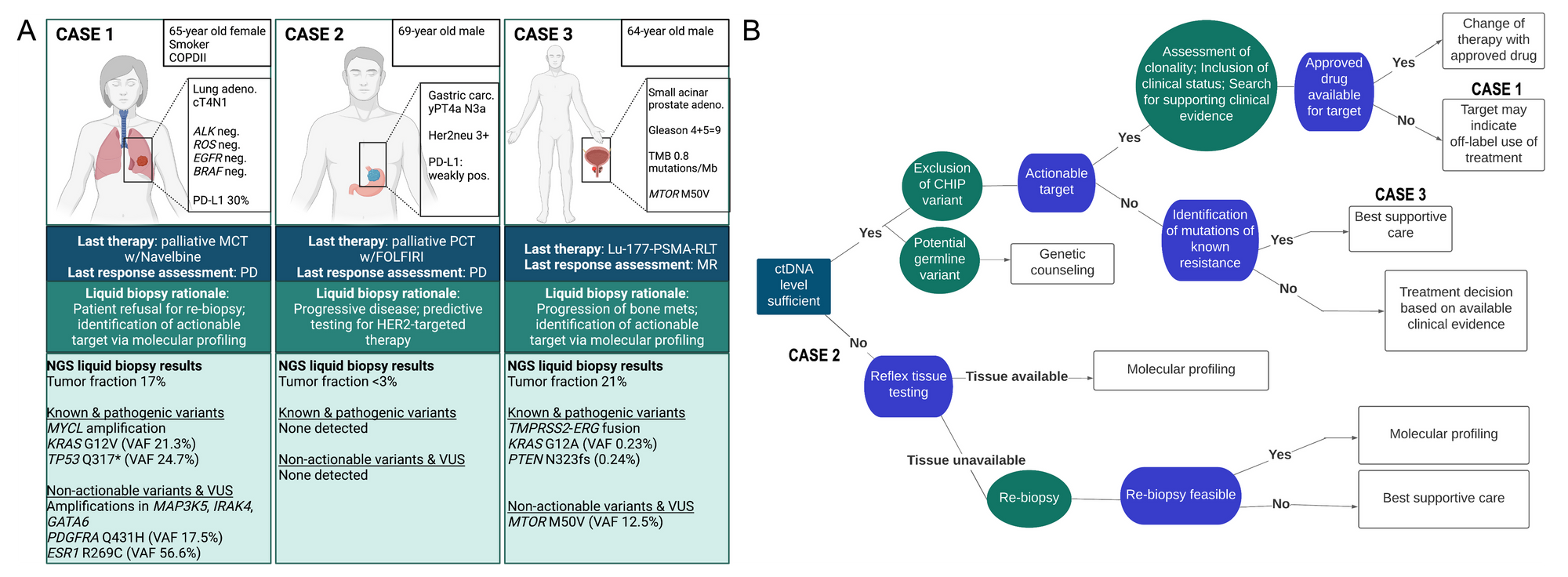
Figure 1. Use cases for ctDNA analysis throughout the cancer patient journey: identification of actionable targets in patients with advanced cancer. A Representation of 3 real-world cases of patients with confirmed progressive disease where liquid biopsy was justified to identify actionable targets. B Basic decision tree for this use case and the interpretation of detected alterations from liquid biopsy NGS data. The cases in (A) are mapped at the corresponding position that reflects the individual scenario.
In the description of Case 1 below, we will describe an overview of the NGS workflow and our methods that apply to all cases presented here so that you have an idea as to how the data were generated.
Case 1
White and dark blue boxes: Without going into too much detail about the patient’s history, we are just going to provide a brief summary. Here we have a case of a 65-year old female with a lung adenocarcinoma who is a current smoker with COPD. At the time of diagnosis, this patient had already presented with multiple metastases, including in the lung and spleen. As is routine, a tissue biopsy of her primary tumor was sent for genomic profiling and did not reveal any alterations in the canonical genes ALK, ROS, EGFR or BRAF. Immunohistochemistry revealed a positive staining for PD-L1 of 30%, for which the patient was later started on nivolumab. After several months of treatment, the tumor progressed and the patient subsequently underwent a series of treatment lines without tumor shrinkage. She was then provided with best supportive care.
Dark green box: Due to further clinical progression, the patient was asked to undergo re-biopsy to obtain more recent tumor tissue for molecular profiling. The patient declined this option and was then offered molecular profiling via liquid biopsy, to which she agreed. In our experience, patients are very willing to donate blood, whereas re-biopsy tends to be met with less willingness.
Light green box: Two vials of blood of approximately 10mL each were sent to us in PAXgene ccfDNA tubes for analysis. After DNA extraction, we employed our standard AVENIO ctDNA Expanded Panel (Roche), which contains 77 genes in the U.S. National Comprehensive Cancer Network (NCCN) Guidelinesas well as emerging cancer biomarkers. As this assay represents comprehensive genomic profiling (CGP), we can detect all four classes of alterations: SNVs, indels, select fusions and select copy number alterations (to see which genes and regions are covered with this panel, see this list of targets).
Just so you have an idea about the duration of the wet and dry lab workflows, here is a rough estimation of the number of hours/days needed:
Table 1. General durations of each step in the wet/dry lab workflows.
Workflow
Step
Duration
Wet lab
DNA isolation
~2 hours
Library preparation
3 days
Library quantification
~2 hours
Sequencing enriched library
~1.5 days
Sequencing whole-genome library
14-17 hours
Dry lab
Secondary data analysis
(variant detection, tumor fraction estimation, copy number calling)
approx. 12 hours
Tertiary data analysis and interpretation
up to 4 hours
This most critical step is the interpretation of the detected variants, which we will touch upon later on.
💡
If you want to see what sample processing, library preparation and sequencing looks like, take a look at our wet lab video tutorials.
Let’s take a look at how we derived the NGS results, which are split up into “Tumor fraction”, “Known and pathogenic variants”, and “Non-actionable variants and VUS”. Here is a breakdown of the main steps to generate the data:
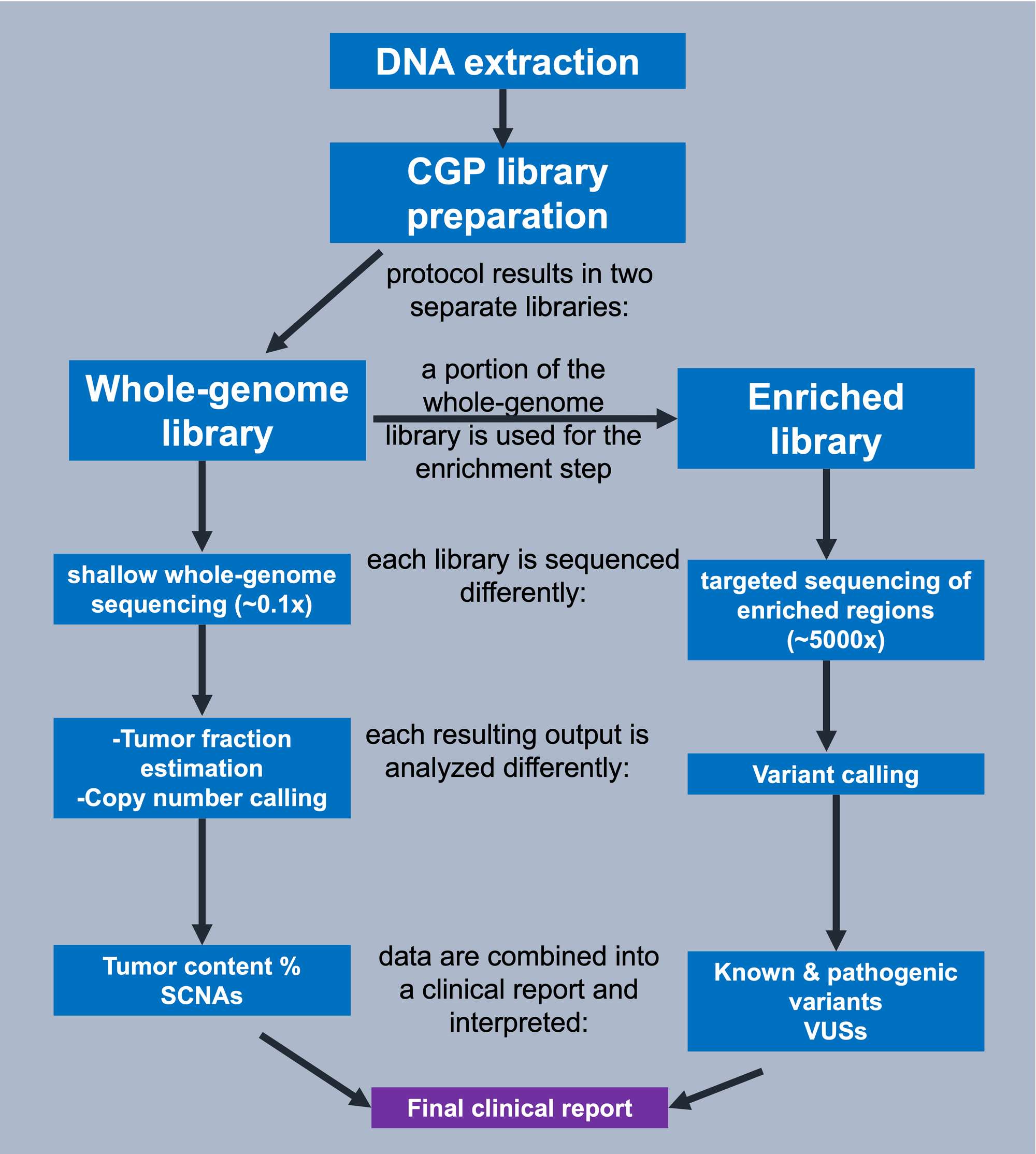
Figure 2.
After DNA extraction from plasma, we begin with comprehensive genomic profiling via the AVENIO ctDNA Expanded panel. This means that using a single assay, i.e. one single DNA source, we can detect the 4 classes of alterations
base substitutions,
insertions and deletions,
somatic copy number alterations (SCNA) and
structural rearrangements.
After adapter ligation and amplification, we can obtain a library of all original cfDNA fragments that were present in the sample, as there was no selection of target regions at this step (here referred to as the whole-genome library). Some of this DNA library is then processed further in the protocol using targeted enrichment. With this hybrid capture-based targeted approach, select regions within the library are captured using long, biotinylated oligonucleotide baits, or probes. These biotinylated baits have been designed to hybridize to regions of interest (here the regions within the 77 genes listed here) within the fragmented cfDNA and streptavidin is subsequently used to separate the baits bound to target DNA from other fragments which were not bound. Each of these libraries, i.e. whole-genome and enriched, are sequenced separately for different purposes. The whole-genome library is sequenced at low coverage, as a shallow coverage of 0.1x suffices for the calling of copy numbers and estimation of tumor fraction. For variant detection, we must sequence the enriched library at high coverage ~5000x in order to ensure reliable variant detection at low allelic frequencies. Once we have our estimated tumor content (provided as a % ), SCNAs of interest, and true variants, we can combine all of this pertinent information into a clinical report and provide an integrated interpretation of the results.
Tumor fraction calculation
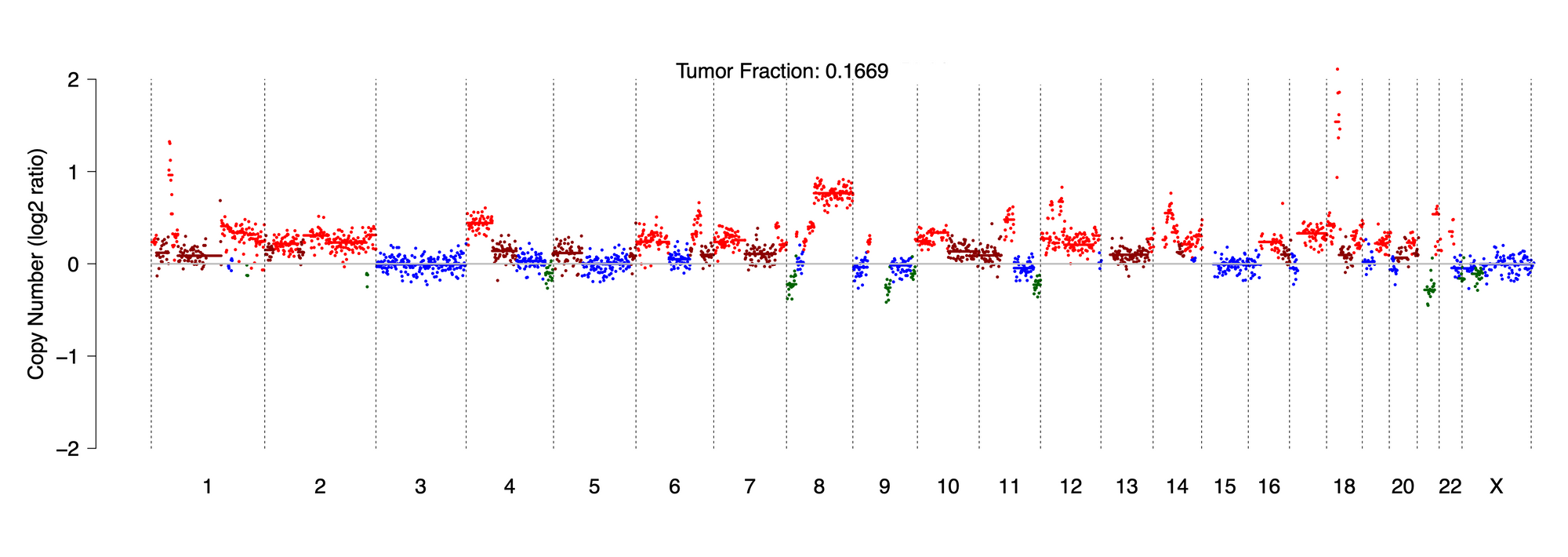
Below, you will learn why the estimation of tumor content in plasma is essential to the interpretation of copy number changes and the variant allele frequencies detected in your sample. To derive tumor fraction, we employ the publicly available ichorCNA algorithm developed and maintained by the Broad Institute. ichorCNA uses a probabilistic model, implemented as a hidden Markov model (HMM), to simultaneously segment the genome, predict large-scale copy number alterations, and estimate the tumor fraction of shallow whole-genome sequencing data. It is optimized for low coverage (~0.1x) sequencing of samples and has been benchmarked using patient and healthy donor cfDNA samples. ichorCNA has a limit of detection of 3%, meaning that liquid biopsy samples with a ctDNA fraction of < 3% indicate lowly detectable levels or the absence of tumor-derived DNA. To run the algorithm, we use our shallow whole-genome sequencing dataset (described in “Known and pathogenic variants”) as an input. Below is the graphical output of the algorithm alongside the tumor fraction estimation for this patient sample in Case 1:
Figure 3. ichorCNA profile depicting genome-wide copy number alterations and estimated tumor fraction
Here you will see a standard copy number plot with the chromosomes on the x-axis and the copy number log2 ratios for each bin in the genome on the y-axis. The color mapping is:
1 copy (i.e. loss) = dark green
2 copies = blue
3 copies (i.e. gain) = brown
4+ copies (i.e. amplification) = red
Notice the high-level amplifications detected on chromosomes 1 and 18. You can see the estimation of tumor content provided as a fraction. Here, we see that the algorithm calculated a tumor content of 16.69%, or roughly 17%. This value is carried over to our final clinical report.
For a more details about data interpretation from ichorCNA, the Github Wiki page provides more information.
Detection of somatic copy number alterations (SCNAs)
See you inside. Members have full access to this document
Your North Star in the Transformation of Precision Oncology
See you inside. Members have full access to this document
Your North Star in the Transformation of Precision Oncology
Consider applying if you want to:
Acquire in-depth knowledge and skills, enabling you to excel in your field.
Continuously improve and stay relevant in your career through regular training and development opportunities, positioning you for career advancement
Connect, network, and collaborate with like-minded professionals, gaining unique insights and opportunities that are only available to members of this community.